ISO 13485

Scientific and Regulatory Significance
Regulatory Convergence
ISO 13485:2016 is recognized the most important regulatory authorities and has been converged with global initiatives such as the Medical Device Single Audit Program (MDSAP). Certification generally facilitates market entry by reducing redundant audits and technical file assessments.
Risk-Based Methodologies
The standard mandates an integrated risk management process, requiring documented, evidence-based evaluations of potential hazards throughout the product life cycle. This encompasses:
- Hazard Identification Enumerating all known and foreseeable hazards.
- Risk Analysis & Evaluation: Estimating and evaluating risk levels under both normal and fault conditions.
- Risk Controls: Implementing safeguards (e.g., design features, protective measures) to reduce risk to acceptable levels.
- Residual Risk Management: Demonstrating that any remaining risks are outweighed by the product’s clinical benefits.
Life-Cycle Approach
The standard covers from conceptual design to post-market activities, ensuring that scientific rigor is applied at every stage. This continuous, cyclical view promotes a closed feedback loop whereby post-market data informs design improvements and corrective actions.
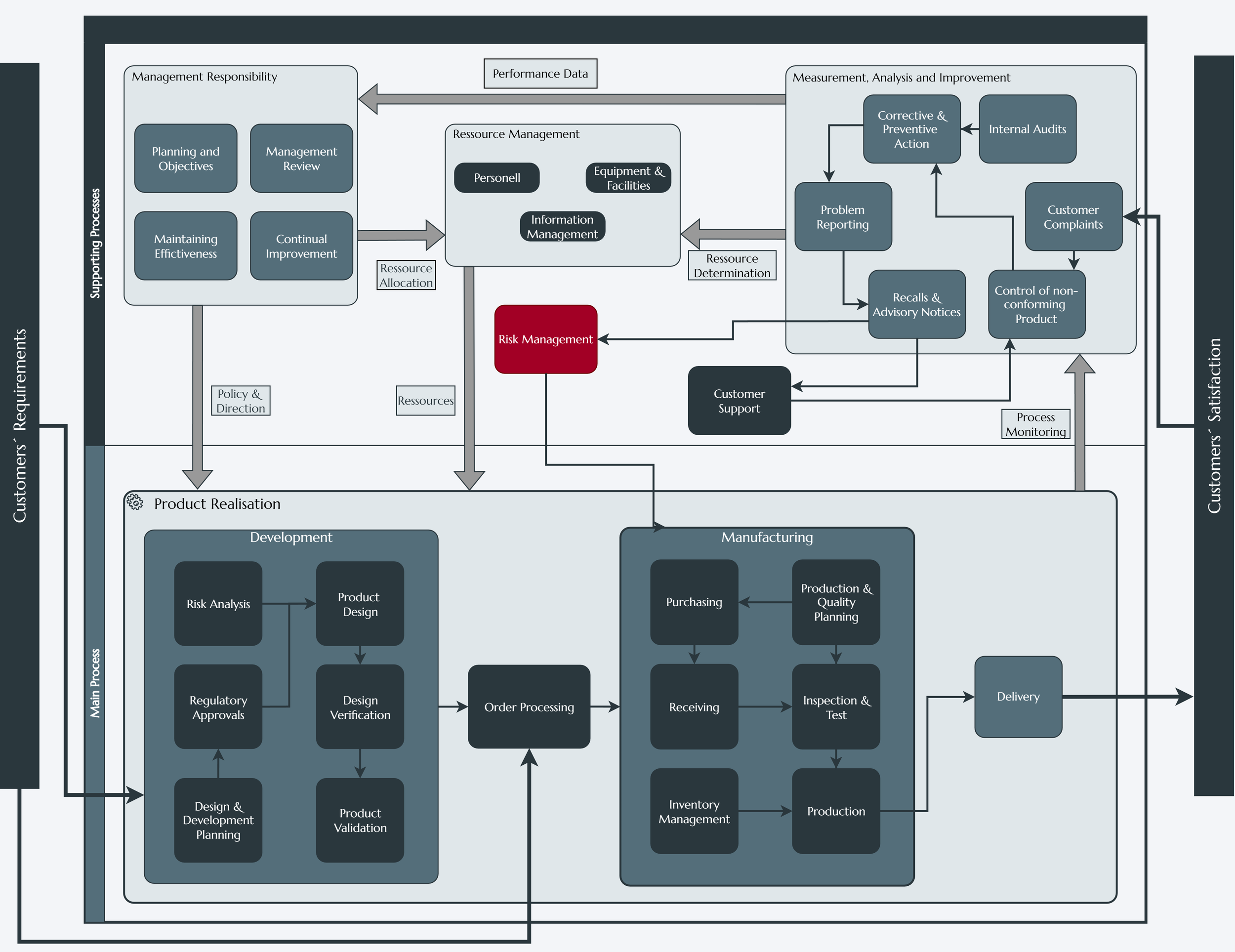
4. Traceability & Product Realization
In a regulated environment, traceability is paramount. ISO 13485:2016 requires meticulous documentation linking each device to its design inputs, components, production parameters, and distribution. These records facilitate prompt investigations, recall actions, or corrective and preventive measures when adverse events are identified.
5. Evidence-Based Continuous Improvement
Data-driven decision-making is integral to ISO 13485:2016. The standard obligates organizations to collect and analyse data on product performance, nonconformities, and customer feedback, using statistical methods where appropriate to detect trends and implement systematic improvements.
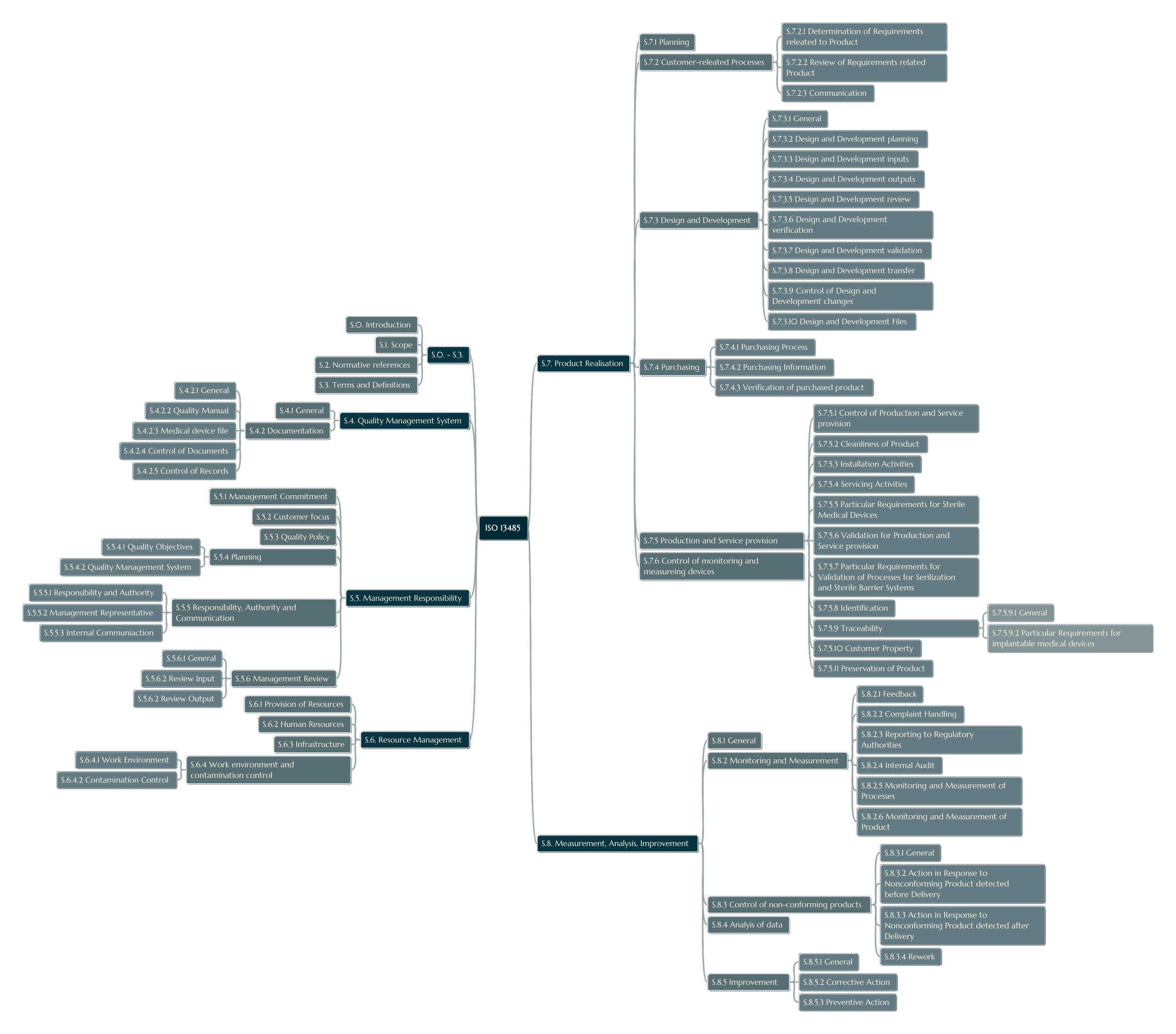
Principal Requirements and Clauses
While ISO 13485:2016 includes a structure akin to ISO 9001, it is adapted with medical device–specific provisions. Some of the most significant clauses include
Quality Management System (Clause 4):
Defines requirements for controlled documentation, computerized system validation, and comprehensive record-keeping—including device files, design dossiers, and production records.
Management Responsibility (Clause 5):
Mandates top management accountability in defining quality objectives, allocating resources, and reviewing the QMS at planned intervals.
Commits to establishing a corporate culture that is scientifically oriented and continually improving the quality.
Resource Management (Clause 6):
Addresses human resources, emphasizing personnel possessing scientific and technical qualifications.
Covers infrastructure elements such as contamination control, precision equipment calibration, and controlled environments for sterile or sensitive products.
Product Realization (Clause 7):
Outlines design and development controls, including verification and validation steps to ensure devices are in line with intended clinical and user requirements.
Imposes rigorous supplier qualification processes and the need for robust monitoring of external providers.
Details instructions for managing production and service operations, particularly processes that require validation (e.g., sterilization, software and hardware deployment or and cleanroom manufacturing).
Measurement, Analysis, and Improvement (Clause 8):
Focuses on monitoring and measuring both processes and products.
Outlines internal audit requirements, corrective and preventive action (CAPA) procedures, and statistical approaches to evaluate product and process performance.
Requires procedures on handling nonconforming products, customer complaints, and implementing continuous improvement practices.
Structured Implementation Pathway
The following diagram illustrates a systematic methodology to adopt ISO 13485:2016. Each step leverages scientific principles —risk assessment, validation protocols, and data
analytics— to foster an evidence-based QMS.
Conduct a systematic comparative assessment between of current operational workflows and ISO 13485:2016 stipulations.
Identify specific technical gaps, such as incomplete / missing risk assessments, deficient design documentation, or inadequate supplier oversight.
Plan & Document
Develop or revise QMS documentation, including standard operating procedures (SOPs), quality manuals, and work instructions that best reflect scientific practices.
Incorporate robust process validation plans for critical operational parameters (e.g., sterilization, device software testing).
Competence & Training
Ensure cross-functional teams —including R&D, manufacturing, and quality control and assurance—possess the requisite technical expertise (e.g., sterilization validation knowledge, biocompatibility tests knowledge, risk analysis methodologies).
Hold periodic competency assessments to maintain and verify up-to-date competencies.
Internal Audits
Systematically evaluate process conformity and performance efficiency.
Use CAPA frameworks to address any nonconformities identified and employ root cause analysis, effectiveness validation, and standard follow-up measures
Certification Audit
Engage a recognized certification body competent to audit the scope of the medical device QMS.
Demonstrate full compliance with Clause 7 (product realization) to prove scientifically validated manufacturing processes and robust traceability systems.
Maintenance & Continuous Improvement
Apply statistical process control (SPC), design of experiments (DoE), and other analytical methods to continuously improve performance.
Update the QMS based on post-market surveillance data, regulatory requirement changes, and advances in medical device science.
ISO 13485:2016 is an internationally harmonized Quality Management System (QMS) standard for organizations involved in the design, production, installation, and servicing of medical devices. Its requirements align with international regulatory expectations (e.g., U.S. FDA’s 21 CFR Part 820, 745) and reflect a strict, risk-based approach to the entire product life cycle.
Key pillars of ISO 13485:2016 include:
Risk Management in accordance with recognized frameworks (e.g., ISO 14971)
Design Controls encompassing the full continuum of design inputs, outputs, verification, validation.
Product Realization via documented and validated manufacturing processes, supplier controls, and traceability measures.
Post-Market Surveillance mechanisms, including complaint handling, vigilance, and continuous improvement.
By adhering to ISO 13485:2016, medical device organizations demonstrate proof of a scientifically grounded commitment to patient safety and product efficacy and performance, strengthening their credibility in highly regulated markets.


ISO 13485:2016
Quality Management with Confidence
Implementing ISO 13485:2016 is a scientifically driven endeavour that enables medical device manufacturers to leverage best-in-class risk management, design controls, and data-driven continuous improvement. The standard’s emphasis on evidence-based decision-making aligns international regulatory regimes and underscores a deep commitment to patient safety and device efficacy.
By partnering with a specialized consultancy, you gain access to:
- Regulatory Expertise: Guidance on aligning ISO 13485:2016 with FDA, EU MDR, MDSAP, and other global requirements.
- Technical Gap Analysis: Identification of required design controls, validation activities, and documentation upgrades.
- Customized Training: Personnel skill development in risk management, process validation, and scientific data analysis.
- Audit Support: Systematic preparation for internal and external certification audits, focused on verifiable compliance.
- Post-Certification Maintenance: Ongoing monitoring of new regulatory requirements to keep your QMS dynamically current.
Elevate your medical device operations by integrating the scientific rigor and regulatory compliance stipulated by ISO 13485:2016. Contact SciReg Consult to initiate a structured implementation roadmap, leveraging specialized technical insights that drive the highest standards of device quality and patient safety.

-
ISO 13485:2016 requires a risk-based framework throughout product development and manufacturing. ISO 14971 is the recognized standard for risk management, stipulating systematic methods for hazard identification, risk evaluation, and control.
-
Validation must demonstrate that critical processes (e.g., sterilization, software control, manufacturing under cleanroom conditions) consistently yield a product meeting predetermined specifications. Scientific evidence (e.g., microbial challenge studies, software verification reports) is essential.
-
Documentation must be comprehensive and scientifically robust, enabling full traceability from design inputs to final release. For instance, all design changes require documented rationales, impact analyses, and updated validation evidence.
-
While widely recognized, ISO 13485:2016 certification is typically part of a broader regulatory submission (e.g., [[CE marking]] in the EU or [[510(k)]] in the U.S.). Nonetheless, it provides a solid foundation to fulfil technical and quality-related portions of device registration dossiers.
-
Continuous improvement entails collecting empirical data on process yields, device performance, and post-market behaviour. Leveraging statistical analysis and root cause techniques refines processes, reduces risk, and enhances product reliability.