
United States – FDA Requirements
Qualification expectations are defined within both pharmaceutical and medical device GMP regulations, both of which apply to ATMP manufacturers depending on the equipment and systems in use:
-
21 CFR Part 211 – Current Good Manufacturing Practice for Finished Pharmaceuticals
- §211.68: Requires calibration and inspection of automated and mechanical equipment.
- §211.100: Mandates validation of any process affecting finished product quality.
- §211.110: Emphasizes validated methods to ensure batch consistency and integrity.
-
21 CFR Part 820 – Quality System Regulation for Medical Devices
- §820.75: Requires validation of manufacturing processes, including qualification of systems and equipment used in production.
-
FDA Guidance for Industry: Process Validation (2011)
- Defines a lifecycle approach: Process Design → Process Qualification (including IQ/OQ/PQ) → Continued Process Verification (CPV).
- Especially relevant for ATMPs, where process variability and small-scale manufacturing necessitate continuous verification strategies.
-
FDA Guidance on Software Validation
- Applicable when software controls or monitors GMP systems, including automated bioreactors, digital cleanroom monitoring, and electronic batch records (EBRs), all widely used in ATMP facilities.
Europe – EMA and EU GMP Requirements
The EudraLex Volume 4 series outlines GMP principles for medicinal products, with Annex 15 specifically addressing Qualification and Validation:
-
Annex 15 – Qualification and Validation
- IQ: Confirms proper installation according to the manufacturer's specifications.
- OQ: Demonstrates operational consistency across the full range of expected conditions.
- PQ: Verifies performance under actual or simulated production conditions.
Key principles include:
- Section 3.2: Qualification should follow a risk-based approach, focusing effort on aspects that directly affect product quality or safety.
- Section 4.1: All qualification activities must be documented, traceable, and justified, with clear linkage to validation efforts.
-
EU MDR 2017/745 & IVDR 2017/746
- Require validation of critical manufacturing processes, including evidence of installation, operation, and performance qualification of systems that impact device safety and performance.
-
ATMP-specific Guidance Documents
- EMA’s Guideline on Human Cell-Based Medicinal Products and Guideline on Quality, Non-Clinical and Clinical Aspects of Gene Therapy Medicinal Products both highlight the need for robust facility and equipment qualification as part of the overall control strategy.
International Standards – ISO Requirements
ISO standards provide a global benchmark for implementing and auditing qualification activities, especially critical for multi-national ATMP developers:
-
ISO 13485:2016
– Medical devices – Quality management systems
- Requires process and equipment validation when verification alone is insufficient.
- Essential for ATMP manufacturers using custom or hybrid device-equipment systems.
-
ISO 14971:2019 – Application of risk management to medical devices
- Supports science- and risk-based qualification in ATMP settings, particularly for novel bioprocessing technologies.
-
ISO 14644 Series – Cleanrooms
- Defines classification and qualification requirements for cleanrooms and controlled environments, a critical aspect for aseptic ATMP production.
-
ISO 11135 / ISO 11137 – Sterilization of healthcare products
- Mandates IQ, OQ, and PQ for sterilization systems, highly relevant for ancillary equipment and materials in ATMPs.
-
ISO 11607-2 – Packaging for terminally sterilized medical devices
- Ensures packaging processes are validated; applies to cryogenic containers, sterile kits, and transport packaging in ATMP logistics.
-
ISO 17664:2017 – Processing of health care products
- Addresses qualification of cleaning, disinfection, and sterilization systems for reusable equipment in ATMP suites.
-
ISO 14155:2020 – Clinical investigations of medical devices
- Relevant when ATMP trials use specialized medical device systems (e.g., point-of-care cell handling devices).
Key Regulatory Expectations for Qualification
-
Risk-Based Approach
- Regulatory bodies stress using a risk-based validation strategy to prioritize qualification efforts on critical-to-quality attributes.
-
Comprehensive Documentation
- All qualification phases (IQ, OQ, PQ) must be pre-approved, executed under controlled conditions, and fully documented with traceable evidence.
-
Change Control
- A formal change control process is required to assess the impact of modifications on qualification status and trigger requalification when necessary.
-
Periodic Review & Requalification
- Systems should undergo periodic review and requalification after significant changes, repairs, or according to scheduled maintenance cycles.
-
Training and Competency
- Personnel involved in qualification must be trained, competent, and authorized, with training records maintained as part of the validation file.
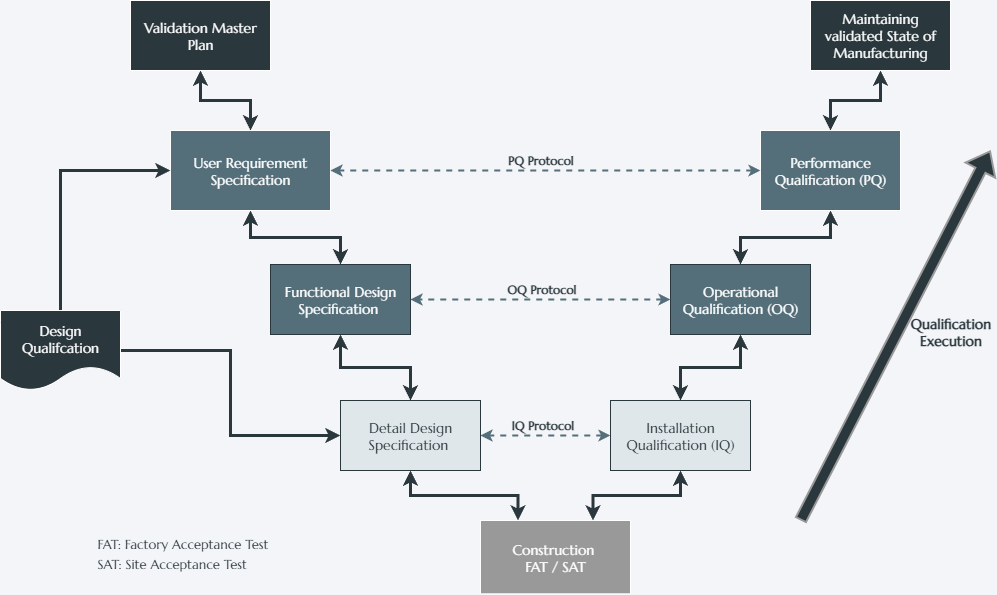
Qualification
Qualification is a cornerstone of Good Manufacturing Practice (GMP) and a critical requirement in Advanced Therapy Medicinal Product (ATMP) manufacturing. It ensures that facilities, utilities, and equipment are properly designed, installed, operated, and maintained so that they consistently deliver products of the highest quality, safety, and efficacy.
In the ATMP setting, where variability in starting materials, small batch sizes, and high patient risk converge, qualification provides the assurance that every critical element of manufacturing is controlled, reproducible, and defensible in front of regulators. Without robust qualification, process validation and ultimately product approval cannot be achieved.
Qualification activities, including Installation Qualification (IQ), Operational Qualification (OQ), and Performance Qualification (PQ), are governed by a globally harmonized framework of regulations and standards. These ensure that manufacturing systems, equipment, and processes are installed, operate, and perform in a manner that guarantees product quality, process control, and patient safety.
Regulatory Framework Overview
Qualification is mandated by leading international regulatory bodies, primarily:
U.S. Food and Drug Administration (FDA)
European Medicines Agency (EMA)
International Organization for Standardization (ISO)
Pharmacopoeias (USP, Ph. Eur.) for specific equipment and cleanroom requirements in ATMP facilities


Make it
Qualification with Confidence
Navigating the complex landscape of global qualification requirements—especially for Advanced Therapy Medicinal Products (ATMPs)—demands more than technical knowledge. It requires a strategic, risk-based approach tailored to your product, process, and facility design.
At SciReg Consult, we translate regulatory expectations into actionable qualification strategies that are practical, compliant, and scalable. Whether you're:
- Constructing a new GMP-compliant cleanroom facility for cell and gene therapies
- Qualifying high-risk, closed-system bioprocessing equipment in aseptic environments
- Aligning your process with global standards such as EU GMP Annex 15, ISO 14644, ISO 13485, and regional ATMP guidelines
We go beyond templated protocols. SciReg delivers:
- Integrated qualification plans: Harmonizing IQ, OQ, and PQ phases with validation master plans and tech transfer timelines.
- GxP-aligned documentation: Tailored for global product submissions and inspections by EMA, FDA, and PMDA.
- Forward-compatible strategies: Supporting future scalability, data integrity, and requalification needs.